Case Study

Iddo Weiner, Converge Bio, Co-founder & CSO
TL;DR. We applied ConvergeCELL™ zero-shot to a held-out bone-marrow single-cell atlas of 84 patients and asked it to distinguish active disease from precursors, surface validated drug targets, and surface disease mechanism. ConvergeCELL™ maps the MGUS → SMM → MM continuum (AUROC 0.72) while two leading baselines drop to chance. It ranks the four immunotherapies specific to MM orders of magnitude higher than any baseline, and recovers the pathway that drives the first-line standard-of-care MM treatment.
Introduction
Multiple myeloma (MM) is a hematological malignancy in which abnormal clones of plasma cells (the antibody-producing arm of the immune system) accumulate in the bone marrow. As these clones expand, they physically crowd the bone marrow and disrupt its normal function, and release a large excess of a single, clonal antibody (the M-protein) into the blood, where it drives the clinical signs of the disease.
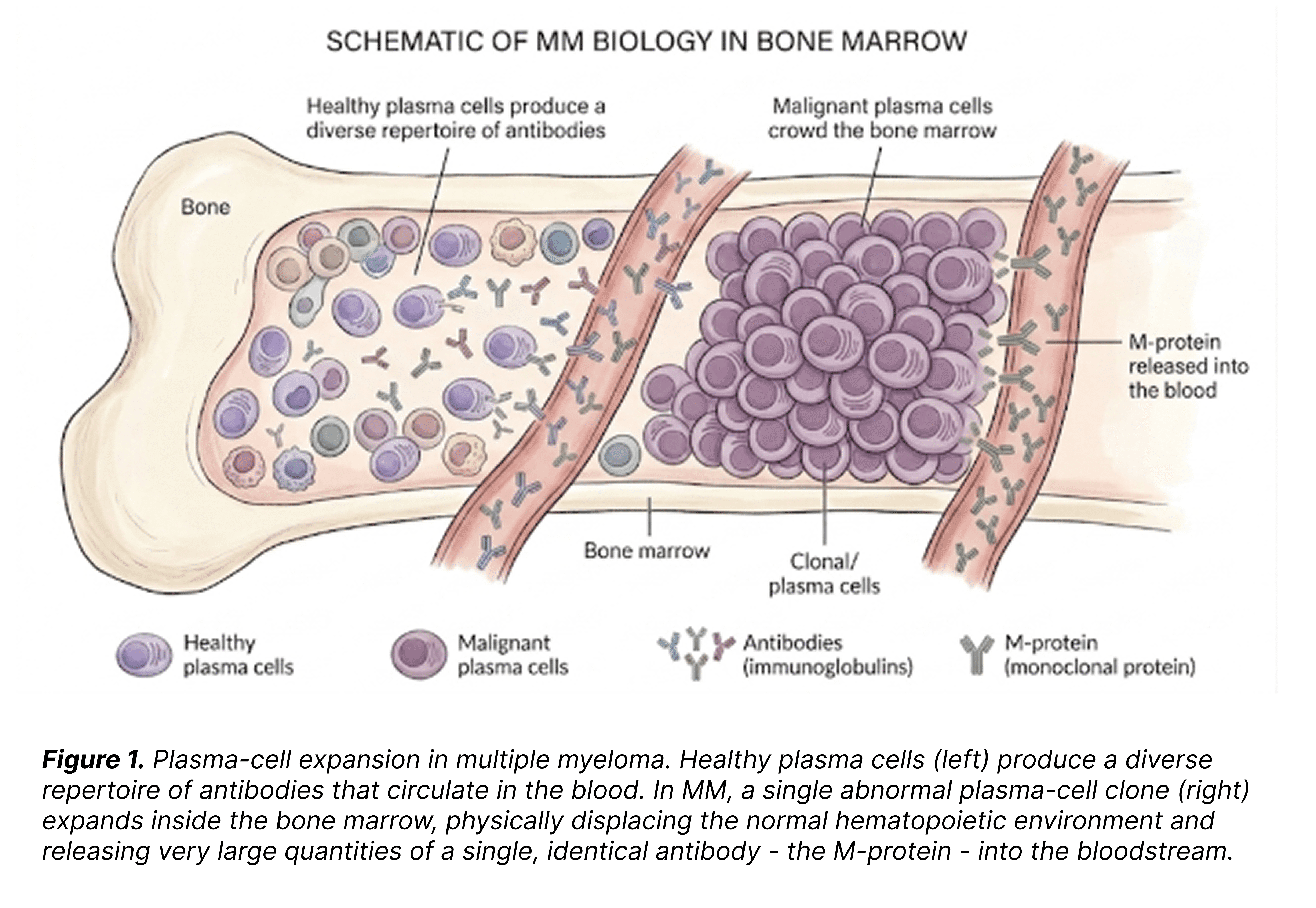
For our purposes here, the relevant feature of MM is its continuous biology. Rather than appearing abruptly as overt disease, MM evolves through a defined sequence of precursor states - monoclonal gammopathy of undetermined significance (MGUS) , smouldering MM (SMM), and active MM. Only the last is considered an active disease.
In terms of disease activity and behaviour, SMM and active MM are essentially identical: the same clonal plasma cells expand, the same M-protein is produced, the same biological process unfolds. The only difference is the clinical presentation - whether that ongoing process has translated into measurable damage to the patient. The four classical clinical signs are known as the CRAB criteria.
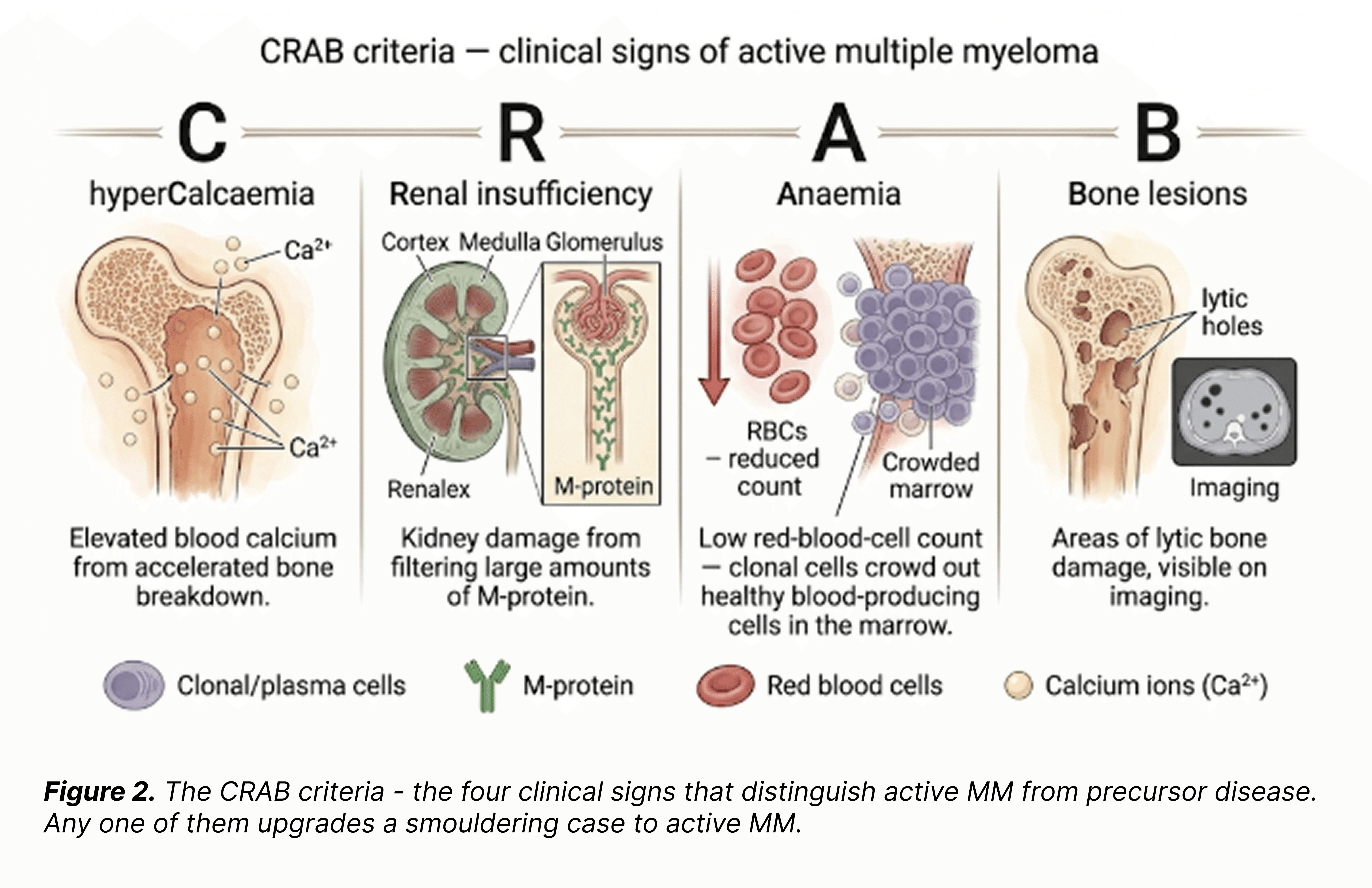
For any model working from RNA alone, the upper end of this continuum is an objectively hard discrimination problem - and a fair test of whether a representation actually captures what the disease is doing.
The case study: what we asked the model to do
Why MM, and what we're really evaluating. This case study is our way of evaluating ConvergeCELL™ as a target-discovery engine. The logic is straightforward: if we apply the model agnostically to a well-studied disease and its top-ranked candidates line up with the clinically validated targets already approved for that disease, that gives us a quantitative basis for trusting the novel candidates it surfaces alongside them - in this disease and in any indication where validated benchmarks are sparse. MM is a good test bed because it carries a substantial set of approved drug targets to check the model's output against.
ConvergeCELL™ is a patient-level single-cell transcriptomics platform pre-trained on 4,479 patients spanning over 350 diseases. The model learns a patient embedding that can be probed for biological structure in several ways: by classifying disease state, by ranking which genes drive the prediction, and by finding which biological pathways those top-ranked genes light up. For this case study, we applied ConvergeCELL™ to a held-out bone-marrow single-cell atlas of 84 patients (Foster et al., Nat Commun 2026) across six independent studies, spanning all three MM stages. We asked the model to do three things at once:
i. Interpret a localized tissue compartment: bone marrow.
ii. Distinguish active disease from precursor stages.
iii. Surface validated and investigational drug targets.
Why we evaluated blind. We deliberately ran ConvergeCELL™ zero-shot: the MM atlas was held out from training, and no MM-specific fine-tuning was applied. This is the hardest test of a pretrained model - what does it already know about a disease the world has not yet told it about? In a partner engagement we would fine-tune on the partner's own cohort and indication; the case study answers the prior question of whether the pretrained model has anything useful to say at all. The numbers below are the floor of what ConvergeCELL can do on this disease, not the ceiling.
Throughout, ConvergeCELL™ is compared against three baselines: pseudobulk differential expression (DE), the standard statistical approach; PaSCient, a published patient-level single-cell foundation model with a native MM disease class; and Standard ML, a PCA-reduced gradient-boosting baseline trained on the same upstream corpus as ConvergeCELL, isolating the contribution of the learned representations.
Result 1: Classification
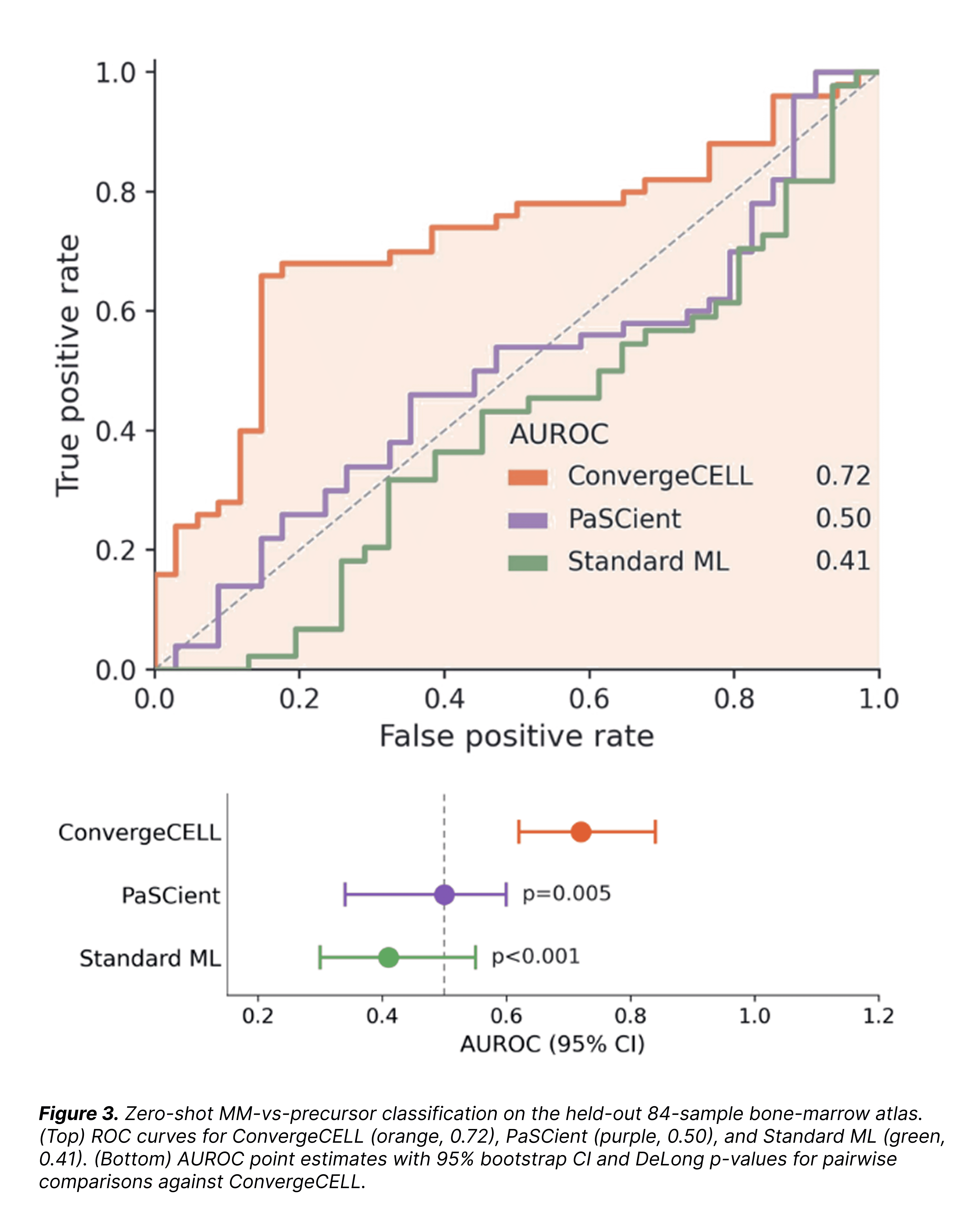
On the binary task of separating active MM from MGUS and SMM precursors, ConvergeCELL™ achieved an AUROC of 0.72. PaSCient, despite having an explicit "multiple myeloma" class in its training labels, landed at AUROC 0.50 (95% CI 0.33–0.60), statistically indistinguishable from a random classifier. Standard ML dropped to 0.41, measurably worse than chance. ConvergeCELL™ is the only method that maps the precursor continuum at all.
The structure of the predictions is more informative than the headline AUROC. ConvergeCELL's confusion matrix shows a graded pattern across the disease arc: MGUS classified as healthy 88% of the time; SMM split roughly 50/50 between healthy and oncological; active MM most strongly oncological (46%), with an immune-inflammatory tail (16%) entirely absent from the precursor stages, consistent with the inflammatory shift that accompanies progression.
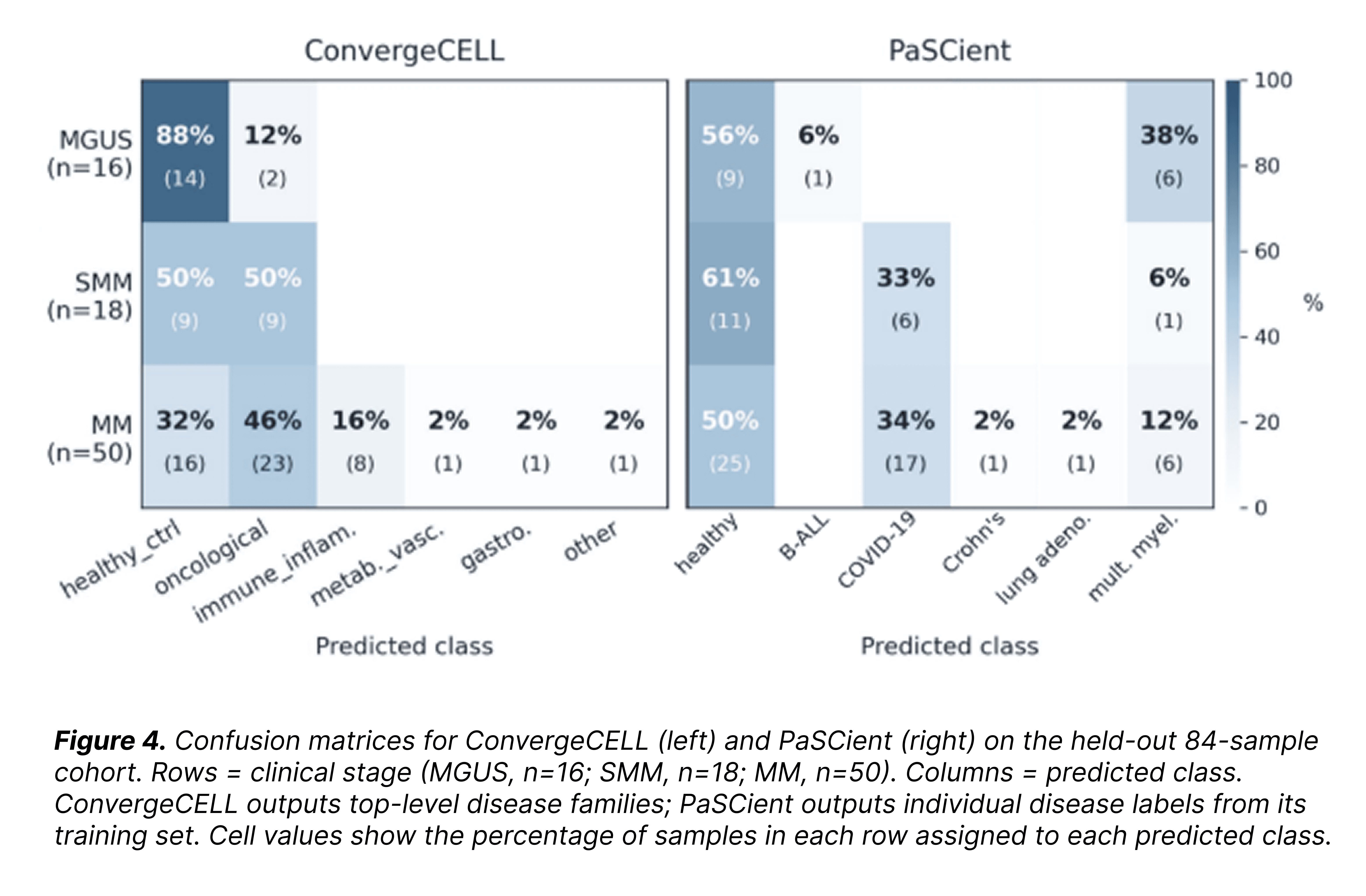
PaSCient, by contrast, assigns half of active-MM samples to "healthy" and a third to "COVID-19", landing only 12% on the correct class even though MM is one of its trained categories. Its AUROC of 0.50 reflects predictions scrambled across the precursor-to-active continuum.
Looking at how ConvergeCELL's predicted oncological probability moves across the disease arc sharpens the picture:
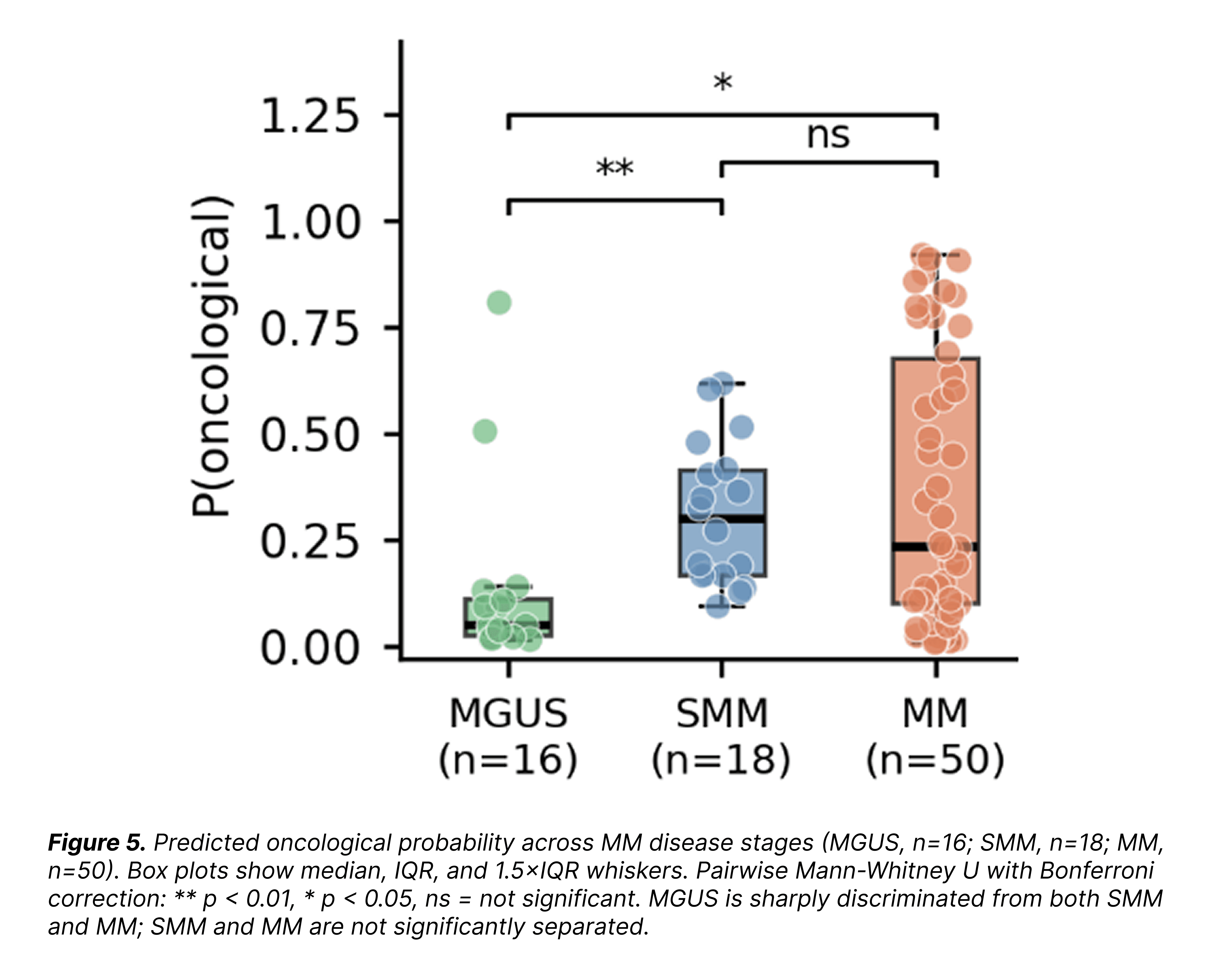
Read together, the AUROC, the confusion matrix, and the probability gradient tell a scientifically honest story. ConvergeCELL™ does separate MGUS from disease. It does not cleanly separate SMM from MM, because the underlying disease activity is the same in both, with only the clinical presentation differing.
Why this matters beyond the AUROC. The same patient embedding that produces these classifications is the substrate we run gene-level attribution on to rank drug targets in the next section. Accurate classification is the foundation of target discovery, wrong classification produces noisy rankings, no matter how good the downstream attribution method.
Result 2: Potential target ranking
Classification is the substrate, not the product. The output for translational use is a ranked list of potential drug targets - genes the model considers most informative about the disease, attributed via integrated gradients on the patient embedding. Many will be novel candidates; some will overlap with already-validated targets. Because MM has a substantial set of established drug targets, we can use those known targets as a reliability benchmark: if the model recovers the targets we already know, that builds confidence in the high-ranked candidates we don't yet know about.
To run this benchmark, we pulled all 10 drugs approved for MM in the Open Targets database, curated each one's primary mechanism-of-action target gene from FDA labeling, and asked each method to place that target somewhere in its ranked list of ~16,000 genes. We grouped the ten drugs into two mechanism classes: disease-specific MM therapies (modern monoclonal antibodies and immunomodulatory drugs targeting plasma-cell biology specifically) and pan-cancer / generic agents (cytotoxic chemotherapies that act on any dividing cell).
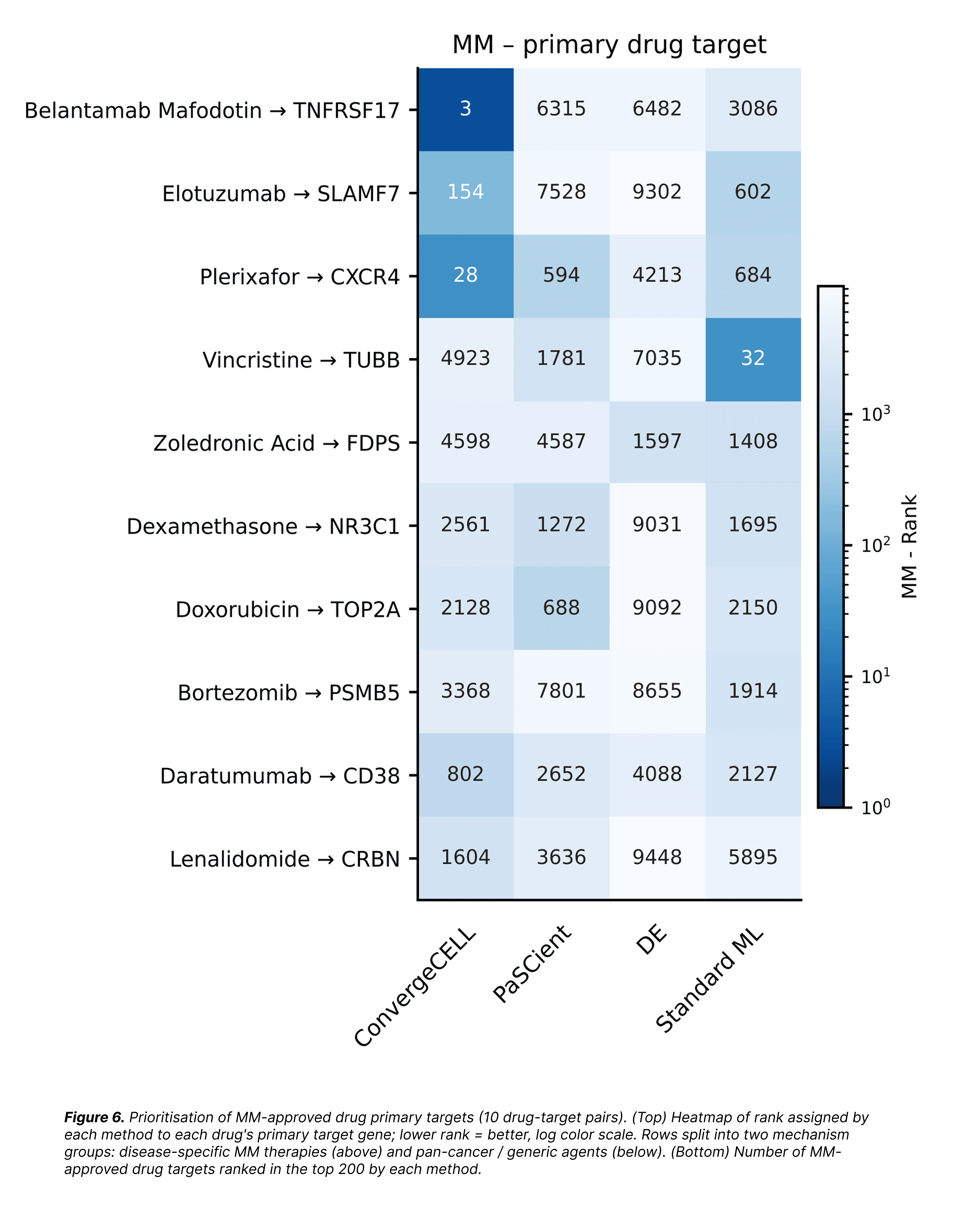
On disease-specific MM therapies, ConvergeCELL™ wins. It ranks three of the four major immunotherapy targets: BCMA (belantamab mafodotin) at rank 3, CXCR4 (plerixafor) at 28, and SLAMF7 (elotuzumab) at 154, within the top 1% of its gene list, blind to their approved status. CD38 (daratumumab) comes in at rank 802, still substantially ahead of every baseline. PaSCient and DE place none of these in the top 200; Standard ML places one, vincristine's pan-cancer (non-specific to MM) cytotoxic TUBB target at rank 32.
On the bottom group of pan-cancer agents, every method performs roughly the same. This is generic biology any method can find. The separation between methods only emerges on targets that require disease-specific representation learning.
Recovering the known disease-specific targets at the top of the ranking is what licenses us to take the rest of the top of the ranking seriously - the novel, uncharacterised, or under-explored candidates that don't yet have an approved drug pointing at them. A target-discovery program isn't ultimately looking for BCMA again; it's looking for whatever the model is ranking just next to BCMA, in the same biological neighbourhood.
Result 3: Pathways
There is one approved-drug class for MM that the gene-level analysis above misses entirely: proteasome inhibitors (bortezomib, carfilzomib, ixazomib), the foundation of first-line MM treatment. PSMB5, bortezomib's primary target, ranks 3,368 by ConvergeCELL™: well outside the actionable zone of any gene-level ranking. No method places PSMB5 highly. Proteasome subunits are constitutively expressed across cell types and aren't differentially regulated in MM vs. precursors. In other words - they are the right drug target for the wrong analytical question.
The right analytical question is not "which gene" but "which biology." We ran pathway-level over-representation analysis against curated Reactome pathways on the top 500 ConvergeCELL-attributed plasma-cell genes, asking which biological program the top-ranked genes collectively belong to.
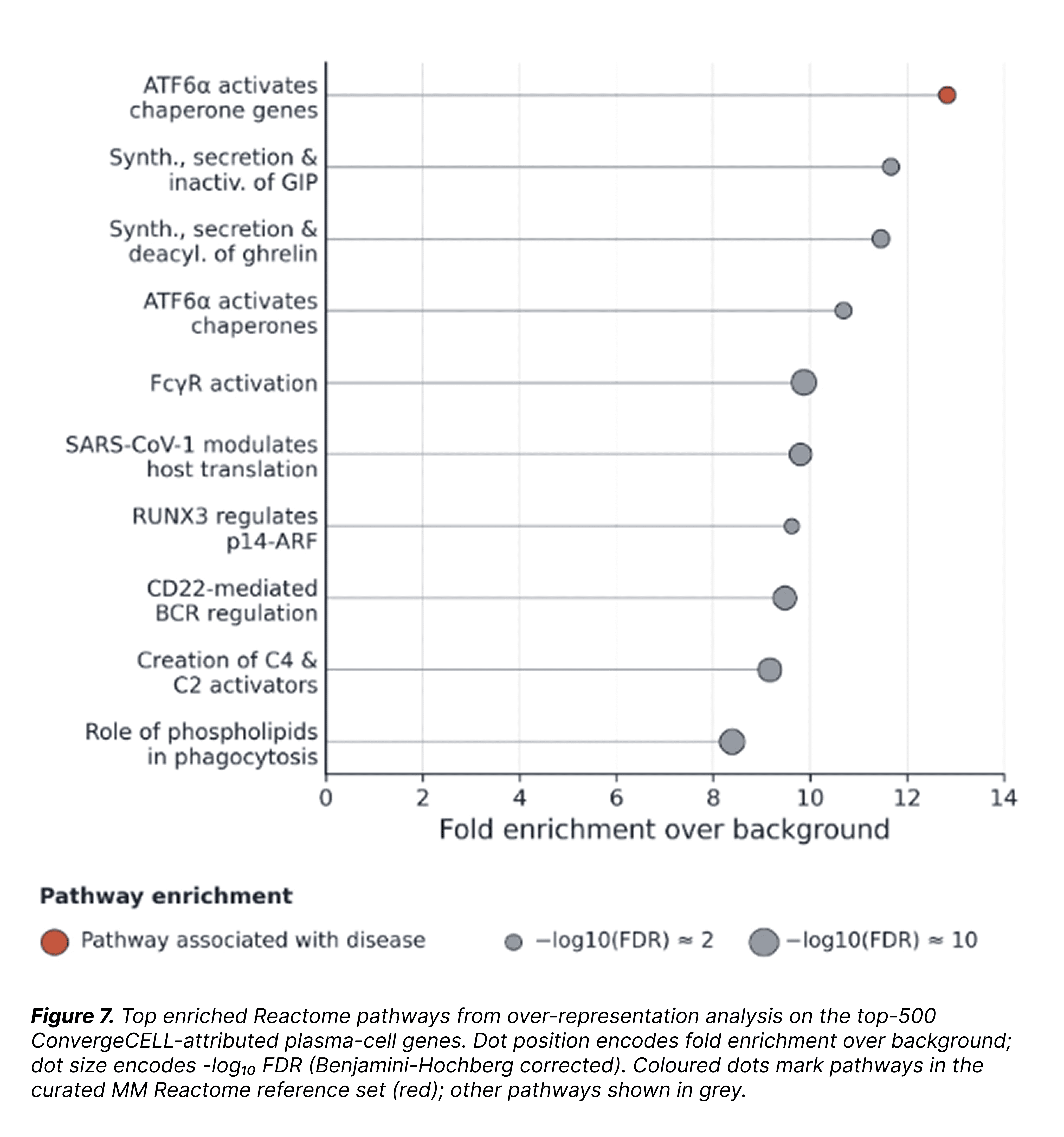
At the top of the enrichment is ATF6α-driven chaperone induction, the canonical branch of the unfolded protein response that plasma cells uniquely depend on to handle their massive antibody-production load. This is precisely the biology proteasome inhibitors exploit: plasma cells already operate close to the ceiling of their proteostatic capacity, and blocking the proteasome pushes them over the edge into apoptosis. ConvergeCELL™ surfaces this dependency from the gene-attribution lists, even though no single proteasome subunit gene ranks high. The signal is in the program, not the gene.
This is the case study's single most important methodological result: one model, two complementary modes of target discovery. Gene-level ranking surfaces individual druggable proteins, antibody, ADC, CAR-T targets that need to be expressed on the surface of the target cell. Pathway-level enrichment surfaces metabolic or stress-response dependencies only visible across the joint behaviour of many genes. The four immunotherapy pillars and the standard-of-care chemotherapy mechanism for MM are recovered together, by the same model, on the same held-out cohort.
Discussion: what this means for partners
1. The model encodes disease biology, not just disease labels.
ConvergeCELL™ recovers the four pillars of MM immunotherapy at the top of its gene-attribution list, blind. That is the consistency a partner needs to trust the novel candidates ranked alongside them: when the model places the targets you already know in the right place, the candidates next to them are much harder to dismiss.
2. Pathway-level discovery is a separate, complementary product.
Most patient-level transcriptomic models are evaluated on gene ranking alone. ConvergeCELL's two-mode discovery: gene ranking for surface targets, pathway enrichment for metabolic and stress-response dependencies, covers two different classes of drug target with the same underlying inference. Where the relevant therapeutic axis is a dependency rather than a surface marker, as with proteasome inhibitors in MM, the gene-level view alone misses the answer.
Conclusion
On a held-out bone-marrow atlas, zero-shot, ConvergeCELL™ maps the MGUS → SMM → MM continuum where standard methods fail; ranks the four pillars of modern MM immunotherapy orders of magnitude higher than any baseline; and surfaces the pathway behind the proteasome-inhibitor backbone of MM standard of care, all from the same patient embedding, on the same cohort.
The results above are what the pretrained model produces out-of-box. For teams running target discovery in any therapeutic area: whether to augment an existing program with novel candidates, de-risk a pipeline by checking what the underlying biology says about the targets you've already prioritised, or surface candidates in indications where validated benchmarks are scarce; ConvergeCELL™ offers a systematic way to extract therapeutic hypotheses from the transcriptomic data your team already has. The case study above is the worst-case scenario: a pretrained model evaluated cold. With fine-tuning on your own cohort and indication, performance only goes up.
Click here to learn more about ConvergeCELL™
Click here to contact the Converge Bio team


