Case Study

Iddo Weiner, Converge Bio, Co-founder & CSO
One Model, Four Targets: Zero-Shot, Generalizable Affinity Maturation with ConvergeAB™
TL; DR.
ConvergeAB™ was applied zero-shot to four antibody–antigen pairs spanning the full antibody optimization continuum, from a discovery-stage hit to an FDA-approved therapeutic. Every target produced improved candidates, with affinity gains of 2.6× to 556× in KD. 18 of 26 measurable wet-lab candidates beat their parent, and developability improved or held on 15 of 20 measurements. Same weights, same procedure, four different biologies, the full optimization continuum.
Introduction
Any single antibody-engineering result invites a fair question: would the same method have worked on a different target? Many platform claims, on closer inspection, were calibrated on a single favourable starting point and never re-run on a target where the answer was not already implicit in the data.
For a partner deciding whether to put a real program through a computational platform, this is the decision that matters. Generalizability, the ability to take an arbitrary starting antibody, against an arbitrary antigen, and reliably deliver an improved molecule without target-specific retraining, is the actual product. A method that works once on a target it was tuned for is a result. A method that works on four targets it was not tuned for, is a platform.
This case study reports a single ConvergeAB™ campaign executed across four starting antibodies in parallel, deliberately chosen to span the range of starting points partners could approach us with: a discovery-stage starting binder (PD-L1, 22.5 nM), a validated binder from a younger antibody class (Spike, 7.5 nM, anti-SARS-CoV-2 RBD), a validated binder from a heavily-engineered antibody class (HER2, 23.9 nM, against a target with multiple approved drugs and decades of intensive antibody engineering), and an FDA-approved therapeutic (tafasitamab, 0.56 nM). Two of the four antigens (PD-L1 and Spike) were also explicitly held out of the model’s training set, so the campaign tests both end-to-end starting-point coverage and out-of-distribution generalization. These four roles probe distinct failure modes: dependence on affinity headroom, over-fitting to training data, brittleness across the optimization spectrum, and CDR-remodelling-driven epitope drift. The campaign was designed so that a method dependent on a favourable starting point would visibly fail on at least one of the four.
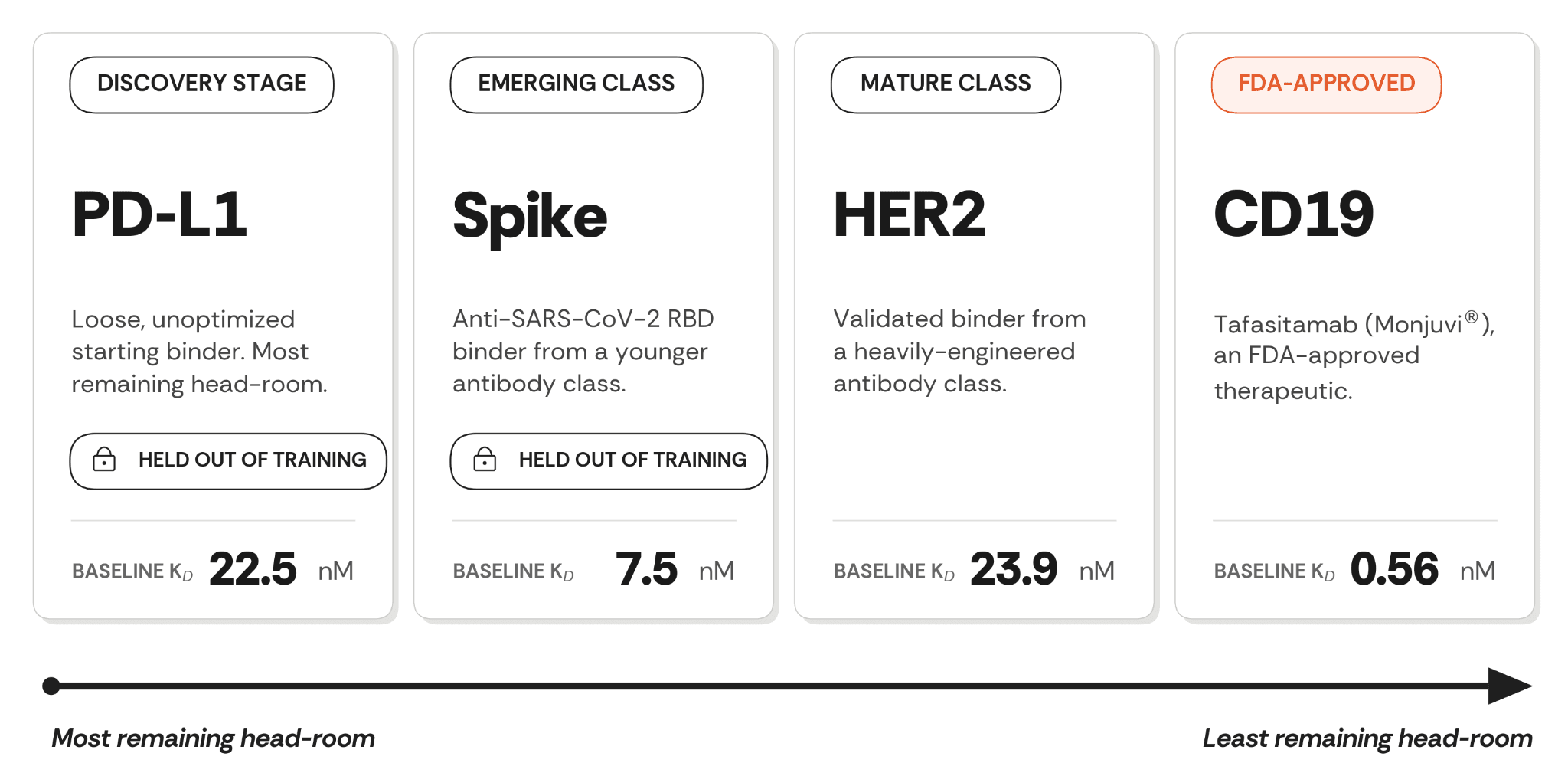
Figure 1. The four starting antibodies and their baselines, ordered along the antibody optimization continuum.
Methods
Design. All four starting antibodies were run zero-shot with ConvergeAB™ from the parent VH/VL sequences and the human target antigen sequence. No target-specific fine-tuning, no per-antibody prompt engineering, no hand-curated training. The same model weights and the same selection procedure were applied to every target. ConvergeAB™ generates sequence diversity around the parent antibody using a proprietary protein language model, producing ~30,000 target-aware candidates per starting antibody, ranked through orthogonal in-silico predictors of binding, affinity, thermal stability, and solubility. The top ~5% advance to atomistic structure simulation and Rosetta-based docking; the resulting ~20 candidates per target are reduced to a diversity-aware shortlist of 10–12 sequences, so the wet lab tests genuinely different molecules rather than near-duplicates. In total, 42 candidates across the four targets were advanced to wet-lab testing.
For this campaign, the platform was configured to preserve the inherited binding interface, a designed-in constraint partners often request when the parent's epitope is part of the asset's value. The platform is fully configurable: partners can pin specific regions or residues as no-change, preserve only certain CDRs, or grant the model unconstrained latitude including complete CDR redesign; the configuration is set per program.
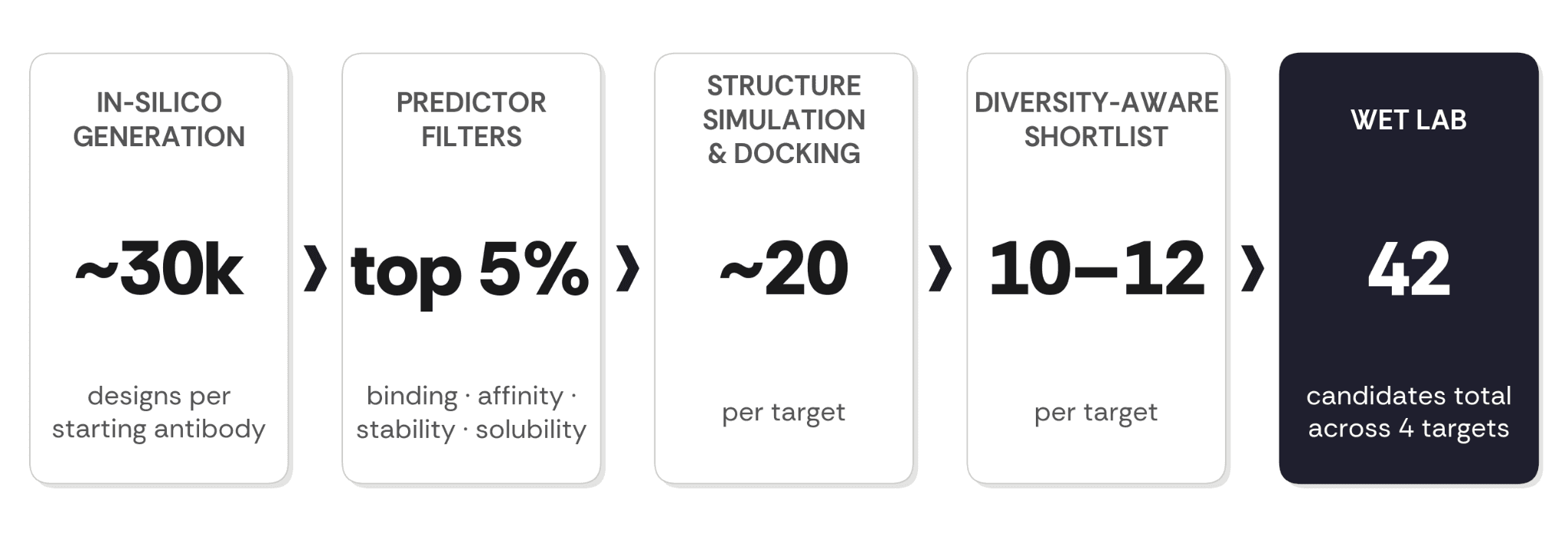
Figure 2. The campaign funnel. Sequence-level predictors filter before structural computation; the final shortlist is diversity-aware.
Experimental panel. All variants were expressed as full-length human IgG1. Binding kinetics were measured by SPR at multiple analyte concentrations to extract kon, koff, and KD. Each candidate was screened against all four intended targets plus IL-23 as an off-target specificity control: five antigens per antibody. Developability was characterized by nanoDSF (Tonset, Tm1), DLS (Tagg), hydrophobic-interaction profiling (HI), and analytical size-exclusion chromatography (% monomer).
Results
Affinity: every target improved
Across the four starting antibodies, every target yielded at least one ConvergeAB-designed candidate with measurably tighter KD than the parent antibody (Table 1).
Target | Position on the optimization continuum | Original KD | ConvergeAB™ lead | Fold ↓ | # AA edits |
|---|---|---|---|---|---|
PD-L1 | Discovery-stage starting binder (antigen held out of training) | 22.5 nM | 0.04 nM |
| 6 |
Spike | Validated binder from a younger antibody class (anti-SARS-CoV-2 RBD; antigen held out of training) | 7.5 nM | 0.29 nM |
| 9 |
HER2 | Validated binder from a heavily-engineered antibody class (decades of HER2 antibody engineering, multiple approved drugs) | 23.9 nM | 9.28 nM |
| 6 |
CD19 | Tafasitamab (Monjuvi®), FDA-approved therapeutic | 0.56 nM | 0.21 nM |
| 10 |
Table 1. Best-of-panel ConvergeAB™ candidate versus parent antibody for each starting antibody. Folds reported on KD. Edit counts are total amino-acid substitutions relative to the parent VH and VL.
Two patterns are worth calling out. First, the magnitude of gain tracks the headroom available for improvement, which is a function of both the starting affinity and the engineering depth of the antibody class. The discovery-stage PD-L1 binder, with a loose starting affinity and ample remaining headroom, produced the campaign’s largest fold-improvement. The Spike binder, tighter but in an antibody class only a few years old, also gained substantially. HER2 illustrates the importance of the second axis: despite a loose starting affinity, it sits in a class with multiple approved drugs and decades of intensive optimization, so its local optimum is close and the gain is smaller but still meaningful. Tafasitamab, already sub-nanomolar after extensive clinical optimization, still delivered a smaller but meaningful improvement. Second, the two largest fold-improvements sit on the two targets whose antigen was held out of the model’s training set (PD-L1 and Spike). The platform has no template in its training corpus for these antibody–antigen pairs; the gains are generated from the model’s sequence-and-structure prior (i.e., its generalized learned knowledge of antibodies), not retrieved from memory.
Figure 3. KD distribution across the wet-lab panel, by target. KD is plotted on a log scale, so lower values indicate tighter binding. The filled orange marker is each target's lead candidate; dark green markers are candidates that beat the parent's KD; open grey circles are candidates whose KD is comparable to or weaker than the parent; grey crosses in the upper band are non-binders shown at the assay limit. The dashed horizontal line marks the parent KD, and the shaded green band beneath it is the improvement zone. Hit counts above each column report improved candidates out of total tested.
Hit rate across the wet-lab panel
Of 42 candidates, 26 produced measurable binding and 18 improved on the parent’s KD (Table 2). Programs with more headroom produced more improved hits; already-optimized parents produced more “comparable” candidates, itself a non-trivial outcome on a tightly-bound parent.
Target | Improved | Comparable | No binding | Panel |
|---|---|---|---|---|
PD-L1 | 7 | 0 | 3 | 10 |
Spike | 5 | 1 | 4 | 10 |
HER2 | 3 | 2 | 5 | 10 |
CD19 (Tafasitamab) | 3 | 5 | 4 | 12 |
Total | 18 | 8 | 16 | 42 |
Table 2. Per-target wet-lab outcomes. "Improved" = measurably tighter KD; "Comparable" = within typical assay variability; "No binding" = no measurable interaction.
Developability: held or improved on 15 of 20 measurements
The four leads were profiled against their parents on five orthogonal developability axes (Table 3). Across 4 × 5 = 20 measurements, 15 improved or held within typical assay variability. The 5 that drifted moved only slightly and remained inside the developable envelope. The developability gains, together with the improved affinity, leave every lead an unambiguous improvement over its parent.
Δ vs parent | Tonset (°C) | Tm1 (°C) | Tagg (°C) | Surface hydrophobicity (lower = better) | % Monomer |
|---|---|---|---|---|---|
PD-L1 |
|
|
|
|
|
Spike |
| −0.6 |
|
|
|
HER2 |
| −3.8 |
| +0.9 | −1.8 |
CD19 (Tafasitamab) |
|
|
| +1.0 |
|
Table 3. Developability deltas of the best ConvergeAB candidate versus its parent, for each starting antibody. Green coloring indicates measurements that improved or held within typical assay variability.
Where the edits land: the model understands biology
The model's edit pattern reflects an understanding of antibody structural biology. When it edits, it preferentially picks positions known to influence CDR-loop conformation: about one-third of the campaign's 31 edits hit canonical Vernier-zone residues (Foote & Winter 1992), framework positions whose side chains physically support and orient the CDR loops. The Vernier zone is a structural definition, a specific set of residues whose role is to tune CDR-loop conformation, independent of which numbering convention is used to label the surrounding regions. A one-third hit rate against a backdrop of ~7% of variable-domain residues being Vernier residues is a ~5-fold enrichment over a uniform-random pick (hypergeometric test, p < 0.0001).
The per-target counts confirm what the affinity results already showed: the model's interventions scale to each parent's remaining optimization headroom. Vernier hits descend the continuum from 4 (PD-L1) to 1 (CD19); CDR edits from 2 to 0. The FDA-approved CD19 (tafasitamab) lead has zero CDR edits, not because the model held back, but because tafasitamab's antigen-contact loops are already where they need to be.
The same structural prior reaches beyond CDR-loop conformation: the edits that drive the affinity gains also drive the developability improvements measured experimentally, suggesting the model has internalized the determinants of thermal stability and surface behavior as well as those of antigen contact. The inherited binding interface is preserved across all four leads, and each lead is a candidate drop-in replacement for its parent in existing assay and formulation pipelines.
Lead | CDR edits | Framework edits | At Vernier-zone positions |
|---|---|---|---|
PD-L1 (discovery-stage) | 2 | 4 | 4 |
Spike (younger class) | 1 | 8 | 3 |
HER2 (mature class) | 1 | 5 | 2 |
CD19 (FDA-approved) | 0 | 10 | 1 |
Table 4. Region-resolved edit counts for each lead. CDR boundaries follow the IMGT convention; Vernier-zone positions per Foote & Winter (1992), defined by structural role. CDR edits and Vernier hits both track the optimization continuum from discovery-stage to FDA-approved.
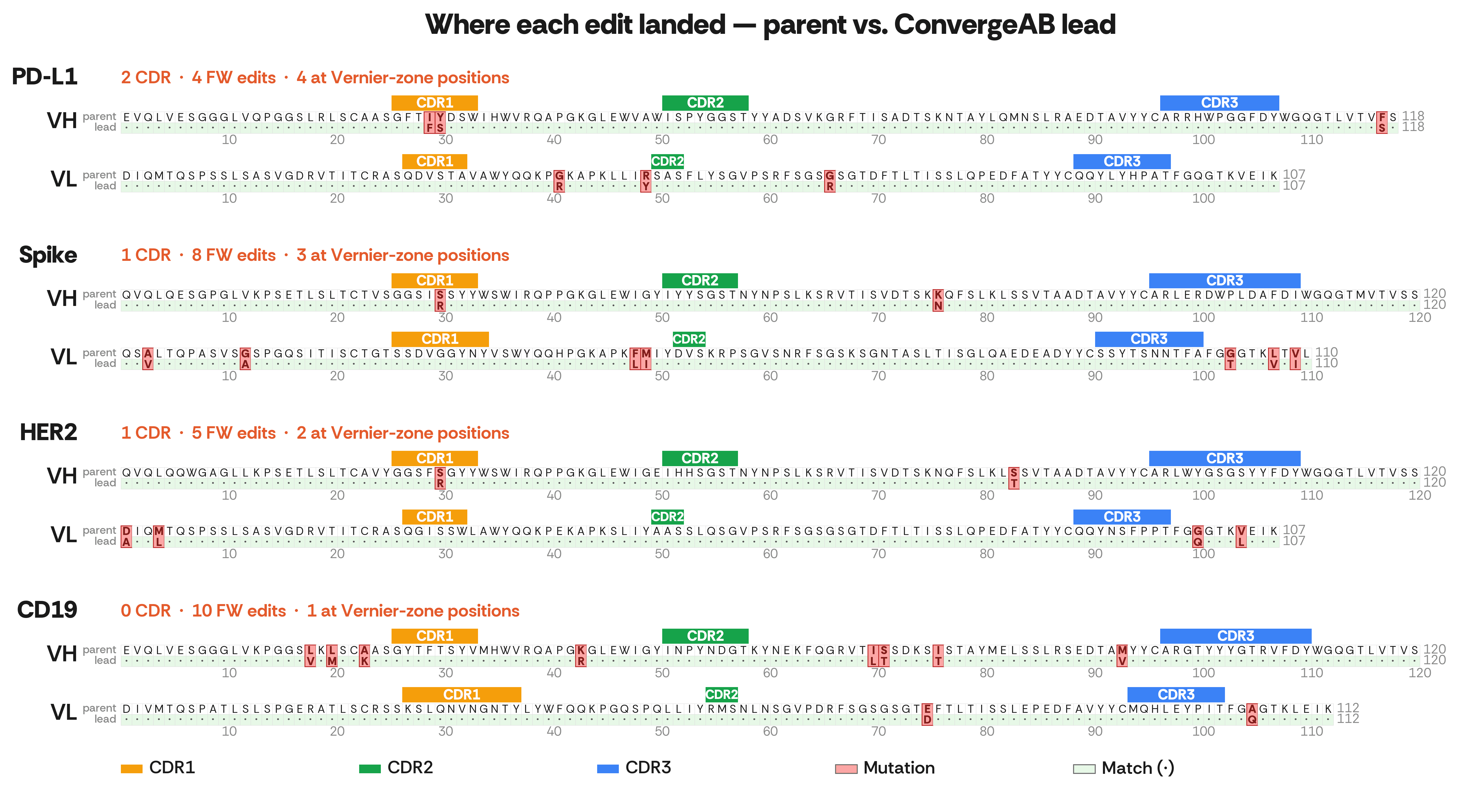
Figure 4. Per-residue alignment between each parent antibody and its ConvergeAB™ lead. Each panel shows the heavy (VH) and light (VL) chains; the top row is the parent residue, the bottom row is the ConvergeAB lead. Matches are shown as dots; mutations are highlighted in pink, spanning both rows. CDR regions are marked with colored bars (CDR1 orange, CDR2 green, CDR3 blue) above the sequence. CDR boundaries follow the IMGT convention. Numbering is sequence position
Discussion: what this means for partners
The four starting antibodies were chosen so that a method dependent on a favourable starting point would visibly fail on at least one of them, and none did. A platform over-fit to parents over-represented in training would have hit the two held-out targets as out-of-distribution walls; instead, Spike and PD-L1 returned the two largest fold-improvements (26× and 556×). A platform dependent on affinity headroom would have returned no improvement on tafasitamab; it delivered 2.7× with developability improved. A platform restricted to modest fold-improvements on loose starting binders would have managed only an incremental gain on PD-L1. Instead, the 556× improvement took a 22.5 nM discovery-stage binder to 0.04 nM in a single design pass, delivering in one round the affinity-maturation outcome partners typically reach only after several iterative campaigns. A platform without configurable region constraints would have been unable to guarantee epitope preservation; the configured paratope-preservation mode applied here delivered minimal, structurally informed edits across all four leads, with the per-lead edit count itself tracking the optimization continuum, and the specificity panel confirms every lead binds only its intended target.
A discovery group does not have the luxury of choosing only programs favourable to a given method; they have the antibody they have, against the antigen they have, often partway through a process. The question is not "what is the best published number" but "what does this engine do on a problem I did not pre-select for it", which is the experiment this campaign was designed to be. The same engine, run zero-shot, delivers across the diversity of antibody starting points partners actually bring to ConvergeAB™, and does so by preserving the binding interface the partner already owns.
Conclusion
Across the four starting antibodies spanning the full antibody optimization continuum, from a discovery-stage starting binder to an FDA-approved therapeutic, ConvergeAB™ improved affinity on every target (2.6× to 556×), preserved or improved developability on 15 of 20 measurements, and beat the parent KD on 18 of 26 measurable candidates. Same weights, same procedure, no per-target fine-tuning. ConvergeAB™ operates as a configurable, target-agnostic optimization engine that delivers value at every point along the optimization continuum, from compressing affinity maturation on discovery-stage hits, to meaningful gains on heavily-engineered antibody classes and FDA-approved drugs, and is positioned to enter partner programs as a drop-in for the binding interfaces they already own.


